| Breast Cancer Gene-Expression Miner v5.0
(bc-GenExMiner v5.0) | |  |
Tutorial - Targeted prognostic analysis
|
|
Targeted prognostic analysis permits to assess the prognostic impact of one gene
or a specific Affymetrix® probeset ID on a selected population.
|
|
|
| Step one |
You have 3 criteria to choose.
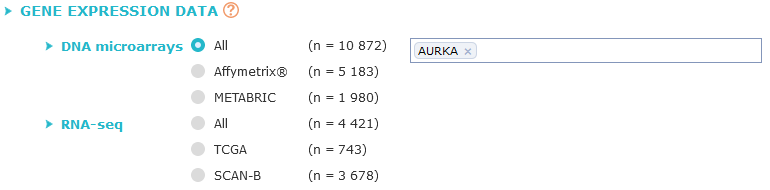
Criterion 1:
gene expression data
First choose the data source (All DNA microarrays, METABRIC, TCGA...), then fill the textbox with actualised* gene symbol (at least 2 characters must be entered)
or
Affymetrix® probeset ID.
A dropdown list will appear, you can then select the gene you want to test.
The list of available genes depends on the previously chosen data source,
if any option, except "Affymetrix®", is checked only gene symbols available with selected data are shown in list.
If "Affymetrix® platform" is checked only gene symbols represented by a probeset are listed.
Each probeset can be selected, if there is more than one probeset, three additional options are available:
- Median probe: median value of all probesets corresponding to the selected gene is taken,
- Highest probe: the probeset having the highest expression level is retained for the analysis (highest median value in a majority of U133P2 and U133A datasets;
in case of ties, decision was based on the total number of patients in the cohorts.),
- JetSet probe: probeset with the highest score given by
JetSet algorithm.
*: see actualised web databases (e.g.:
Ensembl,
GeneCards,
HGNC,
NCBI Gene...)
|
|
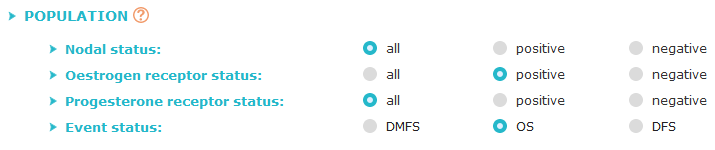
Criterion 2: studied population
Choose the characteristics (nodal, oestrogen receptor, progesterone receptor and event status)
of the cohorts to be explored. "All" status includes patients with any status.
criterion for studied population
Choose the kind of event used for survival analyses:
- "DMFS" as distant metastasis-free survival: first pejorative event represented by distant relapse,
- "OS" as overall survival: first pejorative event represented by death,
- "DFS" as disease-free survival: first pejorative event represented by any relapse or death.
These datasets are retrieved from
annotated
transcriptomic data.
|
|
Criterion 3:
endpoint
Choose the kind of discretisation used for survival analyses as a splitting criterion:
- median,
- tertile,
- quartile,
- optimal: gene or probeset is split according to all percentiles from the 20th to the 80th, with a step of 5,
and the cutoff giving the best p-value (Cox model) is kept,
- customised percentile: choose any percentile from the 20th to the 80th, with a step of 1, to dichotomise the gene.
Once the 3 criteria have been chosen, click on "Submit".
|
Step two |
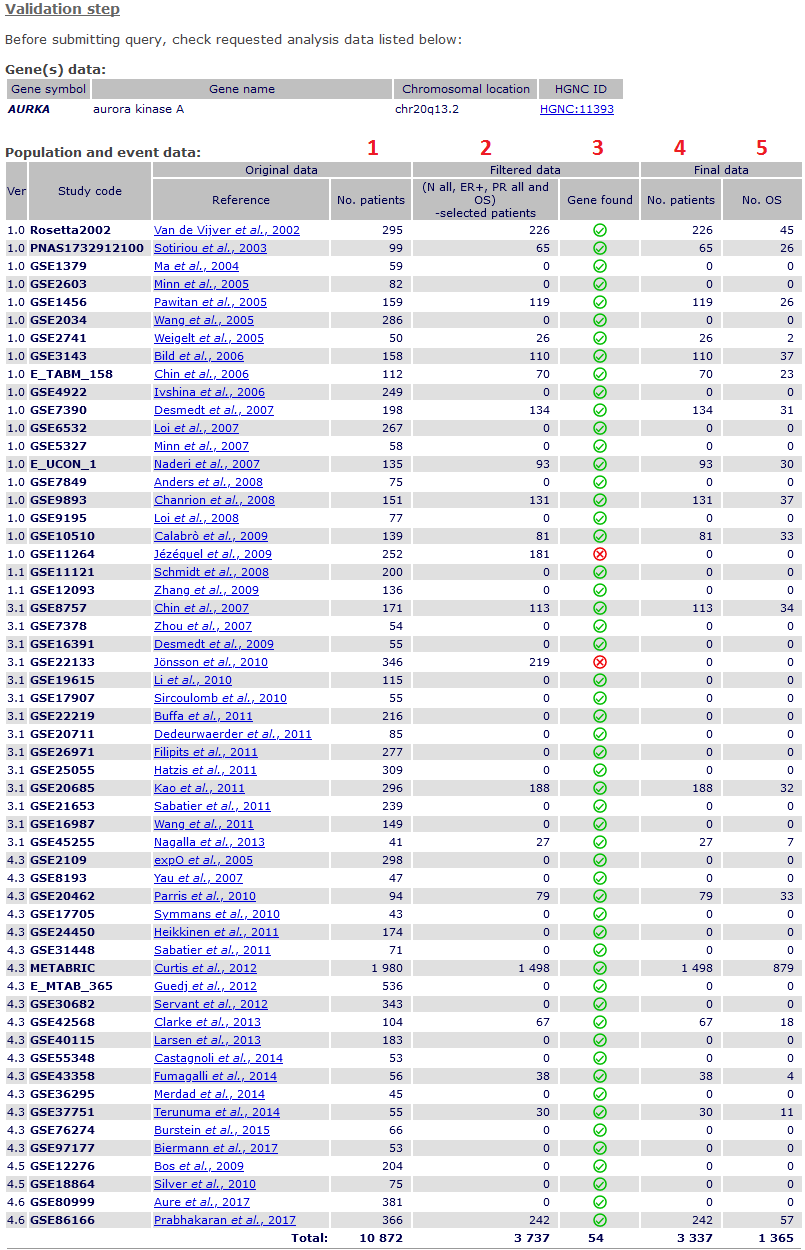
Validation of analysis plan.
After submission, a validation page shows detailed information about:
- tested gene,
- patients from original studies tested,
1 complete data before filtering;
2 results of filtering process;
3 whether the gene has been found;
4 patients and 5 events finally analysed
(if no missing genomic data).
|
|
|
After visualising the validation screen and reading the summary, at the bottom of the page, you can then choose to validate or cancel your submission according
to these intermediate descriptive data summarized at the bottom of the page.
- "Start analysis" will launch
statistical analyses
with the chosen gene and direct you to targeted prognostic analysis result page.
- "Cancel" will redirect you back to previous screen, and offer you to choose a new gene or a new criteria on the form.
|
|
|
|
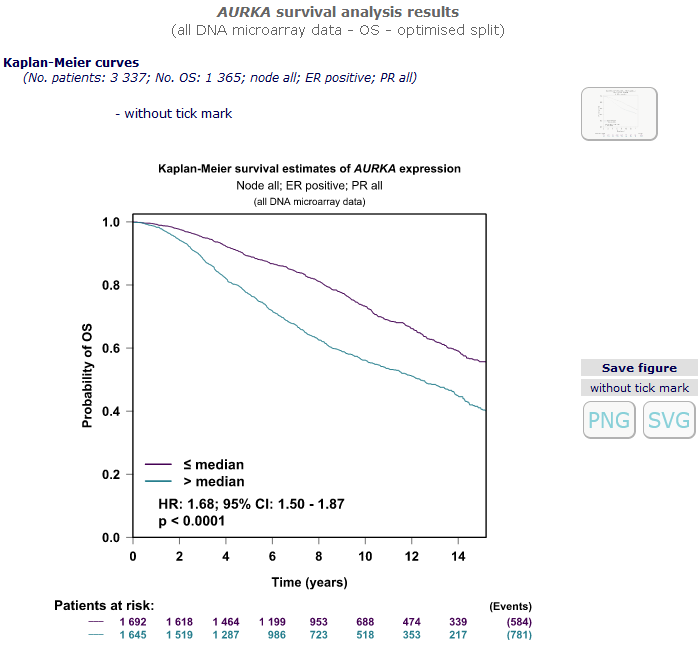
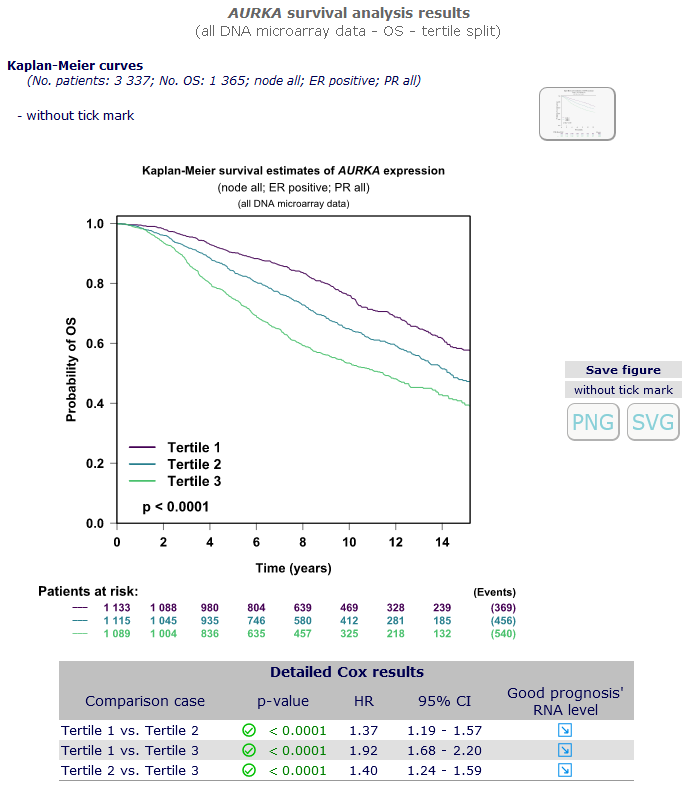
Results are presented in a figure (Kaplan-Meier survival curves) containing univariate Cox scores
(p-values, hazard ratios) for all cohorts of patients fulfilling the chosen criteria, pooled together.
Multivariate Cox scores (adjusted on NPI/AOL/Proliferation score) are also available.
To indicate censored observation both version of the Kaplan-Meier plot is presented: with or without tick marks.
|
|
Detailed results are displayed for the chosen gene by clicking on the appropriate Kaplan-Meier button of your choice on the right side:
Gene's significance for survival is determined in subcohorts of
patients fulfilling the selected criteria. P-value is checked off in green when a
significant link exists (p < 0.05), in orange when there is a trend (p = 0.05-0.10)
or in red when there is no link (p > 0.10).
Moreover the column "Good prognosis' RNA level" shows how the expression level of the gene considered should be in case of good prognosis:
an arrow pointing up indicates that patients with good prognosis have a higher expression level than patients with a bad prognosis;
on the contrary, an arrow pointing down indicates that patients with good prognosis have a lower expression level than patients with bad prognosis.
How to interpret Cox univariate analyses results:
| - |
First, look at p-value: significance is reached when
p-value is below 0.05 and the smallest it is, the best is the prognostic informativity of the gene.
|
| - |
If p-value < 0.05, you then look at the hazard ratio (HR) and its 95% confidence interval (CI):
|
| |
° |
HR is calculated from raw gene's values.
So its value corresponds to the factor by which event-risk is multiplied
when gene raw value increases by 1; a small range of its 95% CI means a good precision.
|
| |
° |
if HR > 1, gene is pejorative and patients
with gene's high values have generally worse prognosis than patients
with gene's low values; the furthest from 1 is the lower bound of the
95% confidence interval, the strongest is the prognostic value of this pejorative gene.
|
| |
° |
if HR < 1, gene is protective and patients
with gene's high values have generally better prognosis than patients
with gene's low values; the furthest from 1 is the higher bound,
the strongest is the prognostic value of this protective gene.
|
| |
° |
Number of patients and especially number
of events used to calculate hazard ratios must also be carefully considered.
When the number of events is 10 or over, robustness of the result is considered strong.
|
Kaplan-Meier
curves permit to visualize results in a different way. Pool's patients are split into groups according to the chosen criterion,
and survival curves are traced for each group.
At the bottom of the graph, number of subjects at risk
are displayed along time of follow-up. In order to minimize unreliability at the end of the curve,
the 15% of patients with the longest follow-up are not plotted.
The Cox model p-value is displayed on the plot.
In case of two groups, relative risk of event (patients with high values / patients with low values) is also displayed
on the plot with its significance and confidence interval.
In case of more than 2 groups, pairwise Cox results are given in a separate table.
Patients with AURKA > pool's optimal split have a risk of metastatic relapse almost x 2 relative to patients
with AURKA < pool's optimal split.
|
Both Kaplan-Meier plots (without or with tick marks) can be saved in "PNG" ([portable network graphics] an universal and easy to use format) or
"SVG" ([scalable vector graphics] a lossless image format figure that allows edit viewing and printing settings, as you wish, for your research article) format. |
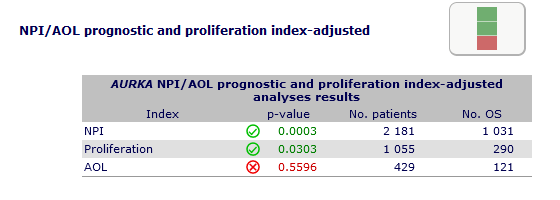
Adjusted analyses
permit to determine if the studied gene bares independent prognostic information over classical
prognostic indexes such as NPI or Adjuvant OnLine and over proliferation score in the pool of cohorts with
NPI, AOL and proliferation score available data. Gene's adjusted HRs are calculated by multivariate Cox.
In this example, it appears that AURKA does not bring additional prognostic information to Adjuvant! but
brings slight additional prognostic information to NPI or proliferation score.
Significant results may be considered robust if Cox tests results are concordant with those results.
Significant results that remain significant in adjusted analyses
would give a supplementary evidence of the gene prognostic interest.
Splitting criteria
that yield more than two groups display one more table below the Kaplan-Meier plot, with the pairwise Cox results.
|
|
|
|