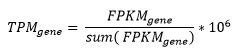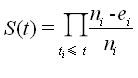| Breast Cancer Gene-Expression Miner v5.0
(bc-GenExMiner v5.0) | |  |
Glossary
[
Published annotated data ][
Published transcriptomic data ][
Intrinsic molecular subtype classification ][
Data pre-processing ]
[
Statistical analyses ][
Survival statistical tests ][
Graphic illustrations ]
Published annotated data:

|
The following inclusion criteria for selection of transcriptomic data were used:
- invasive carcinomas,
- metastasis-free at diagnosis,
- fresh-frozen tumour macrodissection (no microdissection, no formalin-fixed paraffin-embedded, no biopsy [expect for TCGA]),
- no neoadjuvant therapy before tumour collection,
- minimum number of patients per cohorts: 35,
- no duplicate sample inside and between datasets, filtering:
. by sample ID and,
. by a threshold of Pearson correlation ‹ 0.99 which is used to avoid duplicate data,
- female breast cancer.
|
| Ver1 | Reference | No. patients | ER | PR | HER2 | Nodal
status | Histo.
type | PTS | SBR | NPI | AOL | Age diagn. | Ki67 | P53 | BRCA | SSPs | SCMs | IC | Event status | | IHC | seq | GES | DMFS | OS | DFS | | Healthy | | - | - | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | Total for healthy: 0 | | Tumour-adjacent | | - | - | 0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | Total for tumour-adjacent: 0 | | Tumour | | 1.0 | Van de Vijver et al.
2002 | 295 | 295 | 41 | - | 295 | - | - | 41 | 40b | - | 41 | - | - | - | - | - | 295 | 295 | - | 101 | 79 | 122 | | 1.0 | Sotiriou et al.
2003 | 99 | 99 | - | - | 99 | 99 | - | 99 | 99 | 99 | 99 | - | - | - | - | - | 90 | 99 | - | 30 | 45 | 53 | | 1.0 | Ma et al.
2004 | 59 | 59 | 59 | 55 | 52 | 59 | - | 59 | 52 | 52 | 59 | - | - | - | - | - | 59 | 59 | - | - | - | 27 | | 1.0 | Minn et al.
2005 | 82 | 82 | 82 | 76 | 82 | - | - | - | - | - | 82 | - | - | - | - | - | 82 | 82 | - | 27 | - | 27 | | 1.0 | Pawitan et al.
2005 | 159 | 159a | - | - | - | - | - | 147 | - | - | - | - | - | - | 159 | - | 159 | 159 | - | 40 | 40 | 50 | | 1.0 | Wang et al.
2005 | 286 | 286 | - | - | 286 | - | - | - | - | - | - | - | - | - | - | - | 286 | 286 | - | 107 | - | 107 | | 1.0 | Weigelt et al.
2005 | 50 | 50 | - | - | 50 | - | - | 50 | 21b | - | 50 | - | - | - | - | - | 50 | 50 | - | 13 | 10 | 13 | | 1.0 | Bild et al.
2006 | 158 | 158a | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 158 | 158 | - | - | 50 | 50 | | 1.0 | Chin et al.
2006 | 112 | 112 | 112 | 78 | 112 | - | - | 107 | 46b | - | 112 | - | 80 | - | - | - | 99 | 112 | - | 21 | 35 | 42 | | 1.0 | Ivshina et al.
2006 | 249 | 245 | - | - | 240 | - | - | 249 | 159b | - | 249 | - | 247 | - | 249 | - | 249 | 249 | - | - | - | 89 | | 1.0 | Desmedt et al.
2007 | 198 | 198 | - | - | 198 | 184 | - | 196 | 196 | 196 | 198 | - | - | - | - | - | 198 | 198 | - | 62 | 56 | 91 | | 1.0 | Loi et al.
2007 | 267 | 261 | 87 | - | 261 | - | - | 208 | 123b | - | 267 | - | - | - | 267 | - | 266 | 267 | - | 66 | - | 88 | | 1.0 | Minn et al.
2007 | 58 | 58 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 58 | 58 | - | 11 | - | 11 | | 1.0 | Naderi et al.
2007 | 135 | 133 | - | - | 129 | - | - | 134 | 129 | 128 | 135 | - | - | - | - | - | 127 | 135 | - | - | 47 | 65 | | 1.0 | Anders et al.
2008 | 75 | 71 | 70 | - | 75 | 74 | - | 64 | 64 | 61 | 75 | - | - | - | - | - | 75 | 75 | - | 14 | - | 14 | | 1.0 | Chanrion et al.
2008 | 151 | 139 | 139 | - | 146 | 139 | - | 144 | 134 | 124 | 151 | - | - | - | - | - | 139 | 151 | - | 46 | 41 | 55 | | 1.0 | Loi et al.
2008 | 77 | 77 | 77 | - | 77 | - | - | 58 | 30b | - | 77 | - | - | - | 77 | - | 77 | 77 | - | 10 | - | 13 | | 1.0 | Calabrò et al.
2009 | 139 | 136 | 136 | 49 | 103 | - | - | - | - | - | 139 | - | - | - | - | - | 116 | 139 | - | - | 63 | 96 | | 1.0 | Jézéquel et al.
2009 | 252 | 239 | 236 | 203 | 252 | - | - | 252 | 252 | 252 | 252 | - | - | - | - | - | - | - | - | 65 | 47 | 68 | | 1.1 | Schmidt et al.
2008 | 200 | 200a | - | - | 200 | - | - | 200 | 200 | - | - | - | - | - | - | - | 200 | 200 | - | 46 | - | 46 | | 1.1 | Zhang et al.
2009 | 136 | 136 | 136 | - | 136 | - | - | - | - | - | - | - | - | - | - | - | 136 | 136 | - | 20 | - | 20 | | 3.1 | Chin et al.
2007 | 171 | 170 | - | - | 170 | - | - | 170 | 170 | - | 171 | - | - | - | - | - | 152 | 171 | - | 38 | 57 | 56 | | 3.1 | Zhou et al.
2007 | 54 | 54 | - | - | 54 | - | - | - | - | - | 54 | - | - | - | - | - | 54 | 54 | - | 9 | - | 9 | | 3.1 | Desmedt et al.
2009 | 55 | 55 | 55 | 45 | 55 | - | - | 55 | - | - | 55 | - | - | - | 55 | - | 55 | 55 | - | - | - | 55 | | 3.1 | Jönsson et al.
2010 | 346 | 335 | 332 | - | - | - | - | 226 | - | - | - | - | - | - | - | - | 346 | - | - | - | 151 | 151 | | 3.1 | Li et al.
2010 | 115 | 115 | 115 | 115 | 115 | 103 | - | 115 | 64b | - | 115 | - | - | - | 115 | - | 115 | 115 | - | 14 | - | 14 | | 3.1 | Sircoulomb et al.
2010 | 55 | 47 | 47 | 37 | 45 | 33 | - | 47 | - | - | 49 | - | 29 | - | 55 | - | 55 | 55 | - | 17 | - | 17 | | 3.1 | Buffa et al.
2011 | 216 | 216 | - | - | 216 | - | - | 191 | 191 | - | 216 | - | - | - | - | - | 216 | 216 | - | 82 | - | 82 | | 3.1 | Dedeurwaerder et al.
2011 | 85 | 84 | - | 85 | 85 | 85 | - | 85 | 29b | - | 85 | - | - | - | 85 | - | 85 | 85 | - | - | - | 36 | | 3.1 | Filipits et al.
2011 | 277 | 277 | - | 277 | - | - | - | - | - | - | - | - | - | - | - | - | 276 | 277 | - | 58 | - | 58 | | 3.1 | Hatzis et al.
2011 | 309 | 304 | 303 | 309 | 309 | - | - | 286 | - | - | 309 | - | - | - | - | - | 309 | 309 | - | 65 | - | 65 | | 3.1 | Kao et al.
2011 | 296 | 296a | - | - | 296 | - | - | - | - | - | 296 | - | - | - | 296 | - | 296 | 296 | - | 63 | 62 | 73 | | 3.1 | Sabatier et al.
2011 | 239 | 237 | 237 | 224 | 233 | 211 | - | 233 | - | - | 238 | 185 | 175 | - | 239 | - | 239 | 239 | - | - | - | 74 | | 3.1 | Wang et al.
2011 | 149 | 149 | 149 | 149 | 148 | 147 | - | 149 | 148 | - | 149 | - | - | - | - | - | 149 | 149 | - | - | - | 10 | | 3.1 | Kuo et al.
2012 | 51 | 51 | 51 | 51 | 51 | 51 | - | 47 | - | - | 51 | - | - | - | - | - | 51 | 51 | - | 12 | - | 12 | | 3.1 | Nagalla et al.
2013 | 41 | 40 | 38 | 39 | 41 | - | - | 39 | 39 | 36 | 41 | - | - | - | - | - | 41 | 41 | - | 14 | 10 | 14 | | 4.3 | expO et al.
2005 | 298 | 210 | 209 | 198 | 257 | 289 | - | 252 | 39b | - | 298 | - | - | - | 298 | - | 298 | 298 | - | - | - | - | | 4.3 | Yau et al.
2007 | 47 | 47 | 47 | - | 43 | - | - | - | - | - | 47 | - | - | - | - | - | 47 | 47 | - | - | - | - | | 4.3 | Parris et al.
2010 | 94 | 94 | 94 | 94 | 94 | 80 | - | 75 | 75 | - | 93 | - | - | - | - | - | 93 | 94 | - | - | 44 | 45 | | 4.3 | Symmans et al.
2010 | 43 | 43 | - | - | 42 | - | - | - | - | - | - | - | - | - | - | - | 43 | 43 | - | 71 | - | 71 | | 4.3 | Heikkinen et al.
2011 | 174 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 172 | 174 | - | 34 | 27 | 34 | | 4.3 | Sabatier et al.
2011 | 71 | 71 | 71 | 19 | 26 | - | - | - | - | - | 44 | - | - | - | 71 | - | 71 | 71 | - | - | - | - | | 4.3 | Curtis et al.
2012 | 1 980 | 1 937 | 1 980 | 1 980 | 1 980 | 1 830 | - | 1 892 | 1 875 | - | 1 980 | - | - | 1 980 | - | 1 980 | 1 978 | 1 980 | 1 973 | 602 | 1 143 | 1 235 | | 4.3 | Guedj et al.
2012 | 536 | 515 | 514 | 390 | 438 | 427 | - | 517 | - | - | 523 | - | 239 | - | 536 | - | 536 | 536 | - | 119 | - | 119 | | 4.3 | Servant et al.
2012 | 343 | - | - | - | 337 | 318 | - | 339 | - | - | 343 | - | 97 | - | - | - | 343 | 343 | - | 119 | - | 119 | | 4.3 | Clarke et al.
2013 | 104 | 101 | - | - | 104 | - | - | 104 | 45b | - | 104 | - | - | - | 104 | - | 104 | 104 | - | 48 | 35 | 48 | | 4.3 | Larsen et al.
2013 | 183 | 183 | 183 | 183 | - | 169 | - | 157 | - | - | 183 | - | - | - | - | 183 | 182 | 183 | - | - | - | - | | 4.3 | Castagnoli et al.
2014 | 53 | 53 | 53 | 53 | 53 | - | - | 53 | - | - | 53 | - | - | - | - | - | 53 | 53 | - | 23 | - | 23 | | 4.3 | Fumagalli et al.
2014 | 56 | 56 | 56 | 56 | 54 | 52 | - | 56 | 54 | - | 55 | 56 | - | - | 56 | - | 56 | 56 | - | - | - | - | | 4.3 | Merdad et al.
2014 | 45 | 38 | 38 | 38 | - | 40 | - | 38 | - | - | 45 | - | - | - | - | - | 45 | 45 | - | - | - | - | | 4.3 | Terunuma et al.
2014 | 55 | 55 | 12 | 12 | 55 | - | - | 48 | 24b | - | 55 | - | 55 | - | - | - | 55 | 55 | - | - | 19 | 19 | | 4.3 | Burstein et al.
2015 | 66 | 66 | 66 | 49 | - | 64 | - | 47 | - | - | 63 | - | - | - | 66 | - | 66 | 66 | - | - | - | - | | 4.3 | Biermann et al.
2017 | 53 | 52 | 52 | 53 | 42 | - | - | - | - | - | 53 | - | - | - | - | - | 53 | 53 | - | - | - | - | | 4.5 | Bos et al.
2009 | 204 | 56 | 56 | 56 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | 4.5 | Silver et al.
2010 | 75 | 35 | 35 | 35 | - | - | - | - | - | - | - | - | - | - | - | 24 | - | - | - | - | - | - | | 4.5 | Burstein et al.
2015 | 198 | 198 | 198 | 198 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | 4.5 | Jézéquel et al.
2015 | 107 | 107 | 107 | 107 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | 4.5 | Jézéquel et al.
2019 | 131 | 131 | 131 | 131 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | 4.6 | Aure et al.
2017 | 381 | 349 | - | 347 | - | - | - | 356 | - | - | - | - | - | - | - | - | 381 | 381 | - | - | - | - | | 4.6 | Prabhakaran et al.
2017 | 366 | 334 | 333 | 298 | - | - | - | 366 | - | - | - | - | - | - | - | - | 366 | 366 | - | 71 | 103 | 119 | | 5.0 | Tseng et al.
2017 | 56 | 56 | 56 | 56 | 25 | - | 56 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | 5.0 | Romero et al.
2018 | 53 | 53 | 53 | 53 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | | 5.0 | Kim et al.
2020 | 84 | 84 | 84 | 84 | - | - | - | 84 | - | - | 84 | - | - | - | - | - | - | - | - | - | 7 | 7 | | Total for tumour: 11552 | | 11 552 | 10 547 | 6 930 | 6 282 | 8 161 | 4 454 | 56 | 8 035 | 4 298 | 948 | 7 838 | 241 | 922 | 1 980 | 2 728 | 2 187 | 10 300 | 10 046 | 1 973 | 2 138 | 2 171 | 3 712 |
- a ER status determined by means of transcriptomics data (Affymetrix™ probe: 205225_at) in case of a lack of IHC data.
See Kenn et al.
- b NPI score could be computed only for node negative patients
|
[ back ]
Published transcriptomic data:
|
|
The following inclusion criteria for selection of transcriptomic data were used:
- invasive carcinomas,
- metastasis-free at diagnosis,
- fresh-frozen tumour macrodissection (no microdissection, no formalin-fixed paraffin-embedded, no biopsy [expect for TCGA]),
- no neoadjuvant therapy before tumour collection,
- minimum number of patients per cohorts: 35,
- no duplicate sample inside and between datasets, filtering:
. by sample ID and,
. by a threshold of Pearson correlation ‹ 0.99 which is used to avoid duplicate data,
- female breast cancer.
|
[ back ]
Intrinsic molecular subtypes classification:
|
Table 1: Intrinsic molecular subtyping methods
|
| MSP |
No. genes in MSP |
Reference |
Platform correspondence |
R script reference |
Statistics |
Subtypes |
| Sorlie's SSP |
500 |
Sorlie et al, 2003 |
Gene symbols; probes median (if multiple probes for a same gene) |
Weigelt et al, 2010 |
Nearest centroid classifier;
highest correlation coefficient between patient profile and the 5 centroids |
Basal-like,
HER2-E,
Luminal A,
Luminal B,
Normal breast-like |
| Hu's SSP |
306 |
Hu et al, 2006 |
| PAM50 SSP |
50 |
Parker et al, 2009 |
| SCMOD1 |
726 |
Desmedt et al, 2008
Wirapati et al, 2008 |
subtype.cluster function, R package genefu |
Mixture of three gaussians;
use of ESR1, ERBB2 and AURKA modules |
ER-/HER2-,
HER2-E,
ER+/HER2- low proliferation,
ER+/HER2- high proliferation |
| SCMOD2 |
663 |
| SCMGENE |
3 |
|
Table 2: Intrinsic molecular subtyping of 16 854 breast cancer patients
included in bc-GenExMiner v5.0 according to 6 molecular subtype predictors.
A DNA microarrays (n = 11 831). B RNA-seq (n = 5 023).
(RSSPC: robust SSP classification based on patients classified in the same subtype with the three SSPs;
RSCMS: robust SCM classification based on patients classified in the same subtype with the three SCMs;
RIMSPC: robust intrinsic molecular subtype predictors classification based on patients classified in the same subtype with the six MSPs)
|
| A |
| MSP | Basal-like | HER2-E | Luminal A | Luminal B | Normal breast-like | unclassified | | No | % | No | % | No | % | No | % | No | % | No | % | | Sorlie's SSP | 1 636 | 15.0 | 1 313 | 12.0 | 3 257 | 29.8 | 1 250 | 11.4 | 1 454 | 13.3 | 2 013 | 18.5 | | Hu's SSP | 2 510 | 23.0 | 983 | 9.0 | 2 658 | 24.3 | 2 006 | 18.4 | 1 662 | 15.2 | 1 104 | 10.1 | | PAM50 SSP | 2 171 | 19.9 | 1 623 | 14.9 | 3 130 | 28.7 | 2 096 | 19.2 | 1 325 | 12.1 | 578 | 5.2 | | RSSPC | 1 482 | - | 444 | - | 1 631 | - | 404 | - | 709 | - | - | - | |
| MSP | ER-/HER2- | HER2-E | ER+/HER2-
low proliferation | ER+/HER2-
high proliferation | - | unclassified | | No | % | No | % | No | % | No | % | - | - | No | % | | SCMOD1 | 2 067 | 18.9 | 1 372 | 12.6 | 3 382 | 31.0 | 3 037 | 27.8 | - | - | 1 065 | 9.7 | | SCMOD2 | 2 194 | 20.1 | 1 440 | 13.2 | 3 250 | 29.8 | 2 919 | 26.7 | - | - | 1 120 | 10.2 | | SCMGENE | 3 099 | 28.4 | 1 599 | 14.6 | 2 895 | 26.5 | 2 470 | 22.6 | - | - | 860 | 7.9 | | RSCMC | 1 488 | - | 788 | - | 2 031 | - | 1 624 | - | - | - | - | - | | | RIMSPC | 1 227 | - | 267 | - | 915 | - | 265 | - | - | - | - | - | | | B |
| MSP | Basal-like | HER2-E | Luminal A | Luminal B | Normal breast-like | unclassified | | No | % | No | % | No | % | No | % | No | % | No | % | | Sorlie's SSP | 582 | 13.2 | 605 | 13.7 | 1 503 | 34 | 625 | 14.1 | 789 | 17.8 | 317 | 7.2 | | Hu's SSP | 954 | 21.6 | 396 | 9.0 | 1 126 | 25.4 | 935 | 21.1 | 869 | 19.7 | 141 | 3.2 | | PAM50 SSP | 783 | 17.7 | 693 | 15.7 | 1 343 | 30.4 | 966 | 21.9 | 602 | 13.5 | 34 | 0.8 | | RSSPC | 544 | - | 199 | - | 708 | - | 210 | - | 410 | - | - | - | |
| MSP | ER-/HER2- | HER2-E | ER+/HER2-
low proliferation | ER+/HER2-
high proliferation | - | unclassified | | No | % | No | % | No | % | No | % | - | - | No | % | | SCMOD1 | 584 | 13.2 | 343 | 7.8 | 1 877 | 42.4 | 1 617 | 36.6 | - | - | 0 | 0.0 | | SCMOD2 | 617 | 14.0 | 397 | 9.0 | 1 801 | 40.7 | 1 606 | 36.3 | - | - | 0 | 0.0 | | SCMGENE | 616 | 13.9 | 406 | 9.2 | 1 838 | 41.6 | 1 561 | 35.3 | - | - | 0 | 0.0 | | RSCMC | 525 | - | 290 | - | 1 500 | - | 1 209 | - | - | - | - | - | | | RIMSPC | 482 | - | 135 | - | 504 | - | 202 | - | - | - | - | - | | |
Figure 1: Intrinsic molecular subtyping of 16 854 breast cancer patients
included in bc-GenExMiner v5.0 according to 6 intrinsic molecular subtype predictors by comparison of source of data: DNA microarrays (outer circles) vs. RNA-seq (inner circles).
A 3 single sample predictors and the robust SSP classification (intersection).
B 3 subtype clustering models and the robust SCM classification (intersection).
C Robust RIMSPC classification (robust intrinsic molecular subtype predictors classification based on patients classified in the same subtype with the six MSPs).
| A |
| Sorlie's SSP | Hu's SSP | PAM50 SSP | RSSPC | Legend | | | | |
|
Basal-like |
|
HER2-E |
|
Luminal A |
|
Luminal B |
|
Normal breast-like |
|
unclassified |
|
| | B |
| SCMOD1 | SCMOD2 | SCMGENE | RSCMC | Legend | | | | |
|
ER-/HER2- |
|
HER2-E |
|
ER+/HER2- low prolif. |
|
ER+/HER2- high prolif. |
|
| | C |
| | | RIMSPC | Legend | | | | |
|
Basal-like |
|
HER2-E |
|
Luminal A |
|
Luminal B |
|
|
Legend 
|
| MSP: | molecular subtype predictor (SSPs + SCMs) | | No.: | number of patients | | RIMSPC: | robust intrinsic molecular subtype predictors classification based on patients classified in the same subtype with the six molecular subtype predictors (3 SSPs + 3 SCMs) | | RSCMC: | robust SCM classification based on patients classified in the same subtype with the three SCMs | | RSSPC: | robust SSP classification based on patients classified in the same subtype with the three SSPs | | SCM: | Subtype clustering model (SCMOD1, SCMOD2 or SCMGENE) | | SSP: | single sample predictor (Sorlie's, Hu's or PAM50) |
|
|
[ back ]
Data pre-processing:
1 DNA microarrays data
1.1 Affymetrix® pre-processing:
Before being log2-transformed, Affymetrix™ raw CEL data were MAS5.0-normalised (Microarray Affymetrix™ Suite 5.0)
using the Affymetrix Expression Console™,
except for Affymetrix™ Gene 1.0 ST which were pre-processed using robust multiarray analysis (RMA) algorithme
from Affy Bioconductor package a.
1.2 Non-Affymetrix pre-processing:
Data have been downloaded as they were deposited in the public databases.
When patient to reference ratio and its log2-transformation were not already calculated,
we performed the complete process.
1.3 Merging data:
Finally, in order to merge data from all studies and create pooled cohorts,
we converted all studies data, except triple-negative breast cancer (TNBC) subtypes, cohorts to a common scale (median equal to 0
and standard deviation equal to 1 b). For TNBC cohorts, ComBat c method was used.
|
|
2 RNA-seq data
2.1 TCGA pre-processing:
2.1.1 All analyses except nature of tissues:
RNA-Seq dataset were downloaded from the TCGA database (Genomic Data Commons Data Portal).
Alignment was performed using STAR two-pass method, and counts were normalized using the FPKM normalization method d
(see protocol here).
FPKM values were log2-transformed using an offset of 0.1 in order to avoid undefined values.
2.1.2 Nature of the tissue:
To carry out analyses according to the nature of tissue, we used already processed RNA-seq data collected by the TCGA.
TPM values were downloaded from GEO via accession number GSM1536837 (tumour) and GSM1697009 (tumour-adjacent).
As detailed on GEO website, reads were aligned against hg19 and quantified using the Rsubread package e.
FPKM values were obtained with with R open source packages edgeR and limma. TPM normalization from the FPKM values.
Once downloaded, gene expression datasets were log2 transformed using an offset of 1.
2.2 GTEx pre-processing:
We used a dataset that contains gene expression values for healthy tissues (no history of cancer, ie reduction mammoplasty) from the GTEx project.
The FPKM values available from GEO (accession number GSE86354) were initially processed and normalized using Rsubread package e and hg19 as reference genome, as for TCGA.
We converted all FPKM gene expression data to TPM data using the formula below:
An offset of 1 was added to the TPM values prior to log2 transformation.
2.3 SCAN-B (GSE81540) pre-processing:
We used the Sweden Cancerome Analysis Network Breast (SCAN-B) f database.
RNA-seq reads were mapped to the hg19 human genome with tophat2 and normalized in FPKM with cufflinks2 pipeline.
Then log2-transformed with an offset of 0.1.
2.4 Merging data:
Finally, in order to merge all studies data and create pooled cohorts,
we converted studies data to a common scale (median equal to 0
and standard deviation equal to 1 b).
For the analysis of nature of the tissue, standardization is not required since RNA-seq raw reads files from different data sources were processed
and normalized with the Rsubread package e, and aligned to the same reference genome UCSC hg19 with the same pipeline.
For TNBC cohorts, ComBat c method was used.
|
[ back ]
Statistical analyses:
|
Several types of analyses are available: correlation analyses, expression analyses and prognostic analyses,
all of which have different subtypes.
|
|
Correlation analyses
|
|
|
Gene correlation targeted analysis:
Pearson's correlation coefficient is computed with associated p-value for each pair of genes based on eight different populations:
- all patients pooled together,
- patients with positive or negative oestrogen receptor (ER) status,
- patients with positive or negative progesterone receptor (PR) status,
- patients with ER and PR combinations statuses,
- PAM50 molecular subtyped patients,
- RIMSPC molecular subtyped patients,
- basal-like (as defined by PAM50) and triple-negative (as defined by immunohistochemistry [IHC]) patients and the intersection of the 2 latter populations,
- and finally triple-negative breast cancer subtypes patients.
Results are displayed in a correlation map, where each cell corresponds to a pairwise correlation
and is coloured according to the correlation coefficient value, from dark blue (coefficient = -1) to dark red (coefficient = 1).
Pearson's pairwise correlation plots are also computed to illustrate each pairwise correlation.
Gene correlation exhaustive analysis:
Pearson's correlation coefficient is computed, with associated p-value, between the chosen gene
and all other genes that are present in the database, based on eight different populations: see list in "Gene correlation targeted analysis" section.
Genes with correlation above 0.40 in absolute value and with associated p-value less than 0.05 are retained and the genes with best correlation coefficients are displayed
in two different tables: one for the first 50 (or less) positive correlations, one for the first 50 (or less) negative ones.
The lists with all genes fulfilling criteria of correlation coefficient above 0.40 in absolute value and associated p-value less than 0.05 can be downloaded from the results page.
|
|
Gene Ontology analysis:
As a complement to this "screening" analysis, an analysis is performed to find Gene Ontology enrichment terms.
This analysis focuses on significantly under- or over-represented terms present in the list of genes most positively correlated with the chosen gene, including itself,
in the list of genes most negatively correlated with the chosen gene and in the union of these two lists.
For each term of each of the Gene Ontology trees (biological process, molecular function and cellular component), comparison is done between
the number of occurrences of this term in the "target list", i.e. the number of times this term is directly linked to a gene,
and the number of occurrences of this term in the "gene universe" (all of the genes that are expressed in the database) by means of Fisher's exact test.
Terms with associated p-values less than 0.01 are kept.
Gene correlation analysis by chromosomal location:
Pearson's correlation coefficient is computed, with associated p-value,
between the chosen gene and genes located around the chosen gene (up to 15 up and 15 down) on the same chromosome,
based on eight different populations: see list in "Gene correlation targeted analysis" section.
Pearson's pairwise correlation plots
are also performed to illustrate correlation of each gene with the chosen one.
Targeted correlation analysis (TCA):
As a complement, results of gene correlation analysis for genes selected via the "TCA" column can be displayed.
Targeted correlation analysis ("TCA" button), which aims at evaluating the robustness of clusters, is proposed:
correlation analyses are automatically computed between all possible pairs of genes that compose a selected cluster.
|
|
Expression analyses
|
|
|
Targeted expression analysis:
Once the analysis criteria have been chosen (data source, gene / Probe set to be tested, clinical criterion (criteria) to test the gene against),
the distribution of the gene in the available population (all cohorts with availability of required information pooled together)
according to the population splitting criterion (criteria) is illustrated by
box and whisker,
bee swarm,
violin and
raincloud plots.
To assess the significance of the difference in gene distributions in between the different groups, a Welch's test is performed,
as well as Dunnett-Tukey-Kramer's tests when appropriate.
|
|
Exhaustive expression analysis:
Box and whisker,
bee swarm,
violin and
raincloud plots are displayed, along with Welch's (and Dunett-Tukey-Kramer's) tests
for every possible population splitting criteria for a unique gene.
Customised expression analysis:
Similarly to targeted analysis, distribution of a chosen gene is compared in between groups, but here, the groups are defined based on another gene:
the population (all cohorts with both gene values available pooled together) is split according to the expression level(s) of the latter gene.
|
|
Prognostic analyses
|
|
|
Time-to-event endpoints or event:
The Time-to-event endpoints (or event) used for survival analyses are:
- "distant metastasis-free survival" (DMFS): first pejorative event represented by distant relapse,
- "overall survival" (OS): first pejorative event represented by death,
- "disease-free survival" (DFS): first pejorative event represented by any relapse or death.
Targeted prognostic analysis:
Once the analysis criteria have been chosen (data source, gene / Probe Set to be tested,
nodal, oestrogen receptor and progesterone receptor statuses of the cohorts to be explored, event, on which survival analysis will be based, and splitting criterion for the gene),
the prognostic impact of the gene is evaluated on all cohorts pooled by means of univariate
Cox proportional hazards model, stratified by cohort,
and illustrated with a Kaplan-Meier curve.
Cox results are displayed on the curve. In case of more than 2 groups, detailed Cox results (pairwise comparisons) are given in a separate table.
In order to minimize unreliability at the end of the curve, the 15% of patients with the longest follow-up are not plotted a.
To evaluate independent prognostic impact of gene(s) relative to
the well-established clinical markers NPI b and AOL c (10-year overall survival) and to proliferation score d,
adjusted Cox proportional hazards models are performed on pool's patients with available data.
Exhaustive prognostic analysis:
Univariate Cox proportional hazards model and
Kaplan-Meier curves
are performed on each of the 27 possible pools corresponding to every combination of population (nodal, oestrogen receptor and progesterone receptor status)
for each event criteria (DMFS, OS and DFS)
to assess the prognostic impact of the chosen gene / Probe Set, discretised according to the splitting criterion selected.
Results are displayed by event criteria and population, and are ordered by p-value (smallest to largest).
|
|
Molecular subtype prognostic analysis:
Patients are pooled according to their molecular subtypes, based on three single sample predictors (SSPs)
and three subtype clustering models (SCMs), and on three supplementary robust molecular subtype classifications
consisting on the intersections of the 3 SSPs and/or of the 3 SCMs classifications:
only patients with concordant molecular subtype assignment for the 3 SSPs (RSSPC),
for the 3 SCMs (RSCMC), or for all predictors (RIMSPC), are kept. Univariate Cox proportional analysis
and Kaplan-Meier curves are performed after choosing
data source, gene / Probe Set, molecular subtype populations, kind of event and discretised according to the splitting criterion selected.
TNBC/Basal-like prognostic analysis:
Univariate Cox proportional hazards analyses
and Kaplan-Meier curves
are performed, for the chosen gene / Probe Set, discretised according to the splitting criterion selected
for all-event criteria (DMFS, OS and DFS),
on Basal-like (BL) patients (PAM50), on triple-negative breast cancer (TNBC) patients (IHC) and on patients both TNBC and BL.
TNBC subtypes prognostic analysis:
Univariate Cox proportional hazards analyses and
Kaplan-Meier curves
are performed, for the chosen gene / Probe Set, discretised according to the splitting criterion selected
for all-event criteria (DMFS, OS and DFS),
on the four triple-negative breast cancer (TNBC) subtyped patients (IHC):
- LAR: luminal androgen receptor;
- MLIA: mesenchymal-like immune-activated;
- BLIA: basal-like immune-activated;
- BLIS: basal-like immune-suppressed.
More details about TNBC subtypes classification : article under review.
|
Nota bene:
- When working with gene symbols and in case of multiple probesets for
the same gene, probeset value median is taken as unique value for the gene.
- Kaplan-Meier curves will not be computed in populations with less than 5 patients.
|
[ back ]
Statistical tests:
|
Correlation statistical tests
|
|
|
|
Pearson correlation
- The coefficient:
Pearson correlation coefficient, also known as the Pearson's product moment correlation coefficient and denoted by r, measures the linear dependence (correlation)
between two variables (e.g. genes).
It is obtained by the formula r = cov(G1,G2) / (std(G1)*std(G2)),
where cov(G1,G2) is the covariance between the variables G1 and G2 and std denotes the standard deviation of each variable.
r values can vary from -1 to 1. A negative r means that when the first variable increases, the second one decreases,
a postive r means that both variables increase or decrease simultaneously.
The greater the r in absolute value, the stronger the linear dependence between the two variables, with the extreme values of -1 or 1 meaning a perfect linear dependence
between the two variables, in which case, if the two variables are plotted, all data points lie on a line.
|
|
- The associated p-value:
Along with the Pearson correlation coefficient, one can test if this coefficient is different from 0, knowing that the statistic
t = r*√(n-2)/√(1-r2) follows a Student distribution with (n-2) degrees of freedom, n being the number of values.
The p-value associated with the Pearson correlation coefficient permits thus to know if a linear dependence exists between the two variables.
Note that one has to be careful when interpreting p-value associated with Pearson correlation coefficient: a significant p-value means that a linear dependence
exists between two variables but does not mean that this linear dependence is strong; for example, a coefficient of 0.05 with 1600 data points is associated
with a significant p-value (p = 0.046) but one can certainly not conclude that there is a strong linear dependence between the two variables !
|
[ back ]
|
Expression statistical tests
|
|
|
|
Gene-expression comparisons
To evaluate the difference of gene's expression among the different population groups, Welch's test is used in between the groups.
Moreover, when there are at least three different groups and Welch's p-value is significant (indicating that gene's expression
is different in between at least two subpopulations), Dunnett-Tukey-Kramer's test is used for two-by-two comparisons
(this test permits to know the significativity level but does not give a precise p-value).
|
|
Optimal discretisation
In customised analyses, when choosing "optimal" as the splitting criterion for discretisation, gene / Probe Set is split according to
all percentiles from the 20th to the 80th, with a step of 5, and the cutoff giving the best p-value (Welch's test) is kept.
|
[ back ]
|
Prognostic statistical tests
|
|
|
|
Optimal discretisation
In prognostic analyses, when choosing "optimal" as the splitting criterion for discretisation,
gene / Probe Set is split according to
|
|
all percentiles from the 20th to the 80th, with a step of 5, and
the cutoff giving the best p-value (Cox model) is kept.
|
|
|
Cox model
- Aim of the Cox model:
Cox model is a regression model to express the relation between a covariate,
either continuous (e.g. G gene) or ordered discrete (e.g. SBR grade), and the risk
of occurrence of a certain event (e.g. metastatic relapse).
Its simplified formula for G gene can be written as follows:
h(t,g) = h0(t)*exp(ß.g), where h is the hazard function of the event occurrence at time t,
dependent on the value g of G and h0(t) is the positive baseline hazard function,
shared by all patients.
ß is the regression coefficient associated with G, the parameter one wants to evaluate.
- Interpretation of Cox model results:
There are two particularly interesting results when building a Cox model: the p-value
associated with ß, which tells us whether the covariate (e.g. gene) has a significant
impact on the event-free survival (if the p-value is less than a certain threshold,
usually 5%) and the hazard ratio (HR) (equal to exp(ß)), sometimes summed up by its way
(sign of ß).
|
|
The HR, which is really interesting when the p-value is significant,
is actually a risk ratio of an event occurrence between patients with regards
to their relative measurements for the gene under study. To be more specific,
the HR corresponds to the factor by which the risk of occurrence of
the event is multiplied when the risk factor increases by one unit:
h(t,G+1) = h(t,G)*exp(ß).
The "way" of this HR permits therefore to know how the gene will generally affect
the patients event-free survival.
For example, saying that parameter ß associated with the gene G under study is negative
(thus exp(ß) < 1) means that the greater the value of G, the lower the risk of event:
if A and B are two patients such as A's G value gA is greater than B's G value gB,
then one can say that patient A has a lower risk of metastatic relapse than patient B:
gA > gB, ß < 0
⇒ ß.gA < ß.gB
⇒ exp(ß.gA) < exp(ß.gB)
⇒ h0(t)*exp(ß.gA) < h0(t)*exp(ß.gB), that is, h(t, gA) < h(t, gB).
|
Kaplan-Meier curves
- The Kaplan-Meier estimator:
Kaplan-Meier method, also known as the product-limit method, is a non-parametric method
to estimate the survival function S(t) (= Pr(T > t): probability of having a survival
time T longer than time t) of a given population. It is based on the idea that being alive
at time t means being alive just before t and staying alive at t.
Suppose we have a population of n patients, among whom k patients have experienced
an event (metastastic relapse or death for instance) at distinct times
t1 < t2 < ... < tm
(m=k if all events occurred at different times). For each time ti, let ni designs
the number of patients still at risk just before ti, that is patients who have not
yet experienced the event and are not censored, and let ei designs the number of
events that occurred at ti. The event-free survival probability at time ti, S(ti),
is then the probability S(ti-1) of not experiencing the event before time ti
(at time ti-1) multiply by the probability (ni-ei)/ni of not experiencing the event
at time ti (which by definition of ti corresponds to the probability of not experiencing
the event during the interval between ti-1 and ti): S(ti) = S(ti-1) x (ni-ei)/ni.
The Kaplan-Meier estimator of the survival function S(t) is thus the cumulative product:
|
|
- The curve:
The Kaplan-Meier survival curve, i. e. the plot of the survival function, permits to
visualize the evolution of the survival function (estimate). The curve is shaped like
a staircase, with a step corresponding to events at the end of each [ti-1; ti[ interval.
Tick marks on each curve indicate censored observation.
The illustration of the Kaplan-Meier survival estimator by the Kaplan-Meier survival
curve becomes especially interesting when there are different groups of patients
(e.g. according to different treatments or different values of biological markers)
and one wants to compare their relative event-free survival. The different survival
curves are then plotted together and can be visually compared.
The colour palette used for the curve is from R package viridis a,
it permits to keep the colour difference when converted to black and white scale
and is designed to be perceived by readers with the most common form of color blindness.
- Reliability of the estimation:
Caution must be taken concerning the interpretation of the survival curve,
especially at the end of the survival curve: the censored patients induce a loss
of information and reduce the sample size, making the survival curve less reliable;
the end of the curve is obviously particularly affected. For our analyses, in order
to minimize unreliability at the end of the curve, the 15% of patients with
the longest event-free survival or follow-up are not plotted a.
|
[ back ]
Graphic illustrations:
|
Correlation graphic illustrations
|
|
|
|
Correlation map
A correlation map illustrates pairwise correlations among a given group of genes.
A correlation map is a square table where each line and each column represent a gene.
Each cell represents a mathematical relation between two genes and is coloured according to the value of the Pearson correlation coefficient between these two genes,
from dark blue (coefficient = -1) to dark red (coefficient = 1).
Cells from the diagonal of the correlation map represents "interaction" of a gene with itself and are coloured in black.
|
|
Pairwise correlation plot
On a correlation plot, the least-squares regression line is plotted along with the data points to illustrate the correlation between two given genes.
Pairwise correlation hexagonal bins
For hexbin a correlation plots, an R Package with binning and plotting functions for hexagonal bins is used.
|
|
|
Expression graphic illustrations
|
|
|
|
Box and whisker, bee swarm, violin and raincloud plots
Box and whisker plots permit to graphically represent descriptive statistics of a continuous variable (e.g. gene):
the box goes from the lower quartile (Q1) to the upper quartile (Q3), with an horizontal line marking the median.
At the bottom and the top of the box, whisker indicates the distance between the Q1, respectively Q3,
and 1.5 times the interquartile range, that is: Q1-1.5*(Q3-Q1) and Q3+1.5*(Q3-Q1).
Bee swarm is a one-dimensional scatter plot similar to stripchart, except that would-be overlapping points are separated such that each is visible
(package beeswarma).
Violin plot combines the kernel probability density plot and box and whisker plot.
Density curves are plotted symmetrically on both sides of the box and whisker plot.
|
|
Raincloud plot is a combination of split-half violin, raw jittered data points, and box and whisker plot b.
Box and whisker, bee swarm, violin and raincloud plots permit to visually compare distributions of a gene among the different population groups.
|
[ back ]
|
|
|